Femoral vascular closure : Classification
In general, VCDs can be classified as either active closure devices, which include devices that close the arteriotomy site via either suture devices, clips or collagen plug devices, or passive closure devices, which include devices that help with compression such as clamps and assisted or enhanced coagulation and sealants. The classification is shown in Table 3.
ANGIO-SEAL™
The Angio-Seal™ family (Terumo Medical Corporation, Somerset, NJ, USA) consists of three devices - Angio-Seal™ VIP, Angio-Seal™ STS Plus and Angio-Seal™ Evolution. The core principle of the devices involves sealing the arteriotomy site using an anchor on the inside of the artery and a collagen plug on the outside both of which are linked by a suture. Haemostasis is achieved primarily through this mechanical seal and is supplemented by the coagulation-inducing properties of the collagen. All three structures are completely bio-absorbed by 3 months of implantation.
The devices are available in 6Fr and 8Fr configurations.
ANGIO-SEAL™ STS Plus
The Angio-Seal™ STS Plus Platform features a self-tightening suture, which eliminates the need for the post placement spring used in older models ( Figure 2 ). This feature allows the arterial closure procedure to be completed in the catheterisation room. . The device creates a mechanical seal by sandwiching the arteriotomy between a bioabsorbable anchor and a collagen sponge, which dissolves within 60-90 days ( Figure 3 ). Once an Angio-Seal™ is deployed, re-puncture in the same artery is possible within 90 days but must occur either proximal or distal to the deployed device.
The Angio-Seal™ procedure is composed of three stages:
(A) locating the artery;
(B) setting the anchor;
(C) sealing the puncture.
- Insert the arteriotomy locator into the Angio-Seal™ insertion sheath.
- Insert the Angio-Seal™ guidewire into the procedure sheath, which is currently in the patient and remove that sheath, leaving the guidewire in place to maintain vascular access.
- Thread the Angio-Seal™ arteriotomy locator/insertion sheath assembly over the guidewire and insert the assembly into the puncture tract. As the tip of the insertion sheath enters the artery, blood will begin to flow from the drip hole in the locator.
- Slowly withdraw the arteriotomy locator/insertion sheath assembly until blood slows or stops flowing from the drip hole. This indicates that the tip of the Angio-Seal™ insertion sheath just exited the artery.
- From this point, advance the arteriotomy locator/insertion sheath assembly 1–2 cm into the artery until blood begins to flow back again from the drip hole in the locator
- Holding the insertion sheath steady, remove the arteriotomy locator and guidewire from the insertion sheath by flexing the arteriotomy locator at the sheath hub.
- Carefully grasp the Angio-Seal™ device at the bypass tube. Cradle the Angio-Seal™ carrier tube in the palm of the hand and, with the reference indicator facing up, slowly insert the bypass tube and carrier tube into the insertion sheath haemostatic valve.
- Confirm that the reference indicator on the insertion sheath is facing up. This should align with the reference indicator on the device cap. The sheath cap and the device sleeve will snap together when properly fitted.
- With one hand continue to hold the insertion sheath cap steady, while with the other hand grasp the device cap and slowly and carefully pull back. Continue pulling on the device cap until resistance from the anchor catching on the distal tip of the insertion sheath is felt.
- After the anchor position is confirmed by proper alignment of the device cap within the coloured band, while maintaining a grip on the insertion sheath, pull the device cap into the full rear locked position. The device sleeve colour band should now be completely visible.
- Once the previous steps have been performed correctly, slowly and carefully withdraw the device/sheath assembly along the angle of the puncture tract to position the anchor against the vessel wall.
- When the insertion sheath clears the skin, a tamper tube will appear. At this stage, grip the tamper tube and gently advance the collagen while maintaining tension on the suture.
- Continue to withdraw the insertion sheath and device until the clear stop on the suture appears. Continue to pull until the entire suture has been deployed (approximately 4.5 cm beyond the clear stop). The suture will then lock within the device cap where it is attached.
- Maintain tension on the suture. Advance the collagen again with the tamper tube until resistance is felt and the black compaction marker is revealed.
- Cut the suture below the clear stop. Remove the tamper tube using a slight twirling upward motion.
- Gently pull up on the suture and cut it below skin level and below the black compaction marker.
ANGIO-SEAL™ VIP
The Angio-Seal™ VIP device was released onto the international market in 2006. Due to a V-Twist Integrated Platform technology, this device provides a larger collagen footprint. The device features a Bondek® Plus Suture (Teleflex Medical, Limerick, PA, USA) with a patented Polyglyd™ coating (Teleflex Medical) for enhanced lubricity. The lubricious suture enables smooth compaction for a uniform and secure seal, while maintaining secure knot locking ability. It is deployed using the same technique as for the STS system.
ANGIO-SEALTM EVOLUTION
Consistent with its Angio-Seal™ predecessors, Angio-Seal™ Evolution achieves haemostasis through the deployment of an anchor, suture and collagen seal ( Figure 5 ). With Evolution, however, single-handed deployment is possible, which enables physicians to support the puncture site more easily. The device's automated collagen compaction system ensures consistent compaction of the collagen against the exterior wall of the vessel reducing procedural variables.
Trial data for the Angio-Seal™
The Angio-Seal™ pivotal randomised, controlled, multicentre trial was published in 1995 and proved the device was safe and efficacious in a group of 435 patients randomised in a 1:1 fashion to the Angio-Seal device or manual compression [1818. Kussmaul WG, Buchbinder M, Whitlow PL, Aker UT, Heuser RR, King SB, et al. Rapid arterial hemostasis and decreased access site complications after cardiac catheterization and angioplasty: Results of a randomized trial of a novel hemostatic device. Journal of the American College of Cardiology. 1995;25(7):1685-92. ]. Time to haemostasis was considerably shorter (2.5 versus 15.3 minutes, p<0.0001) with a lower complication rate. Subgroup analysis showed benefit in those patients undergoing interventional procedures.
Use of the Angio-Seal has been associated with high success rates in the range of 91-100% [1818. Kussmaul WG, Buchbinder M, Whitlow PL, Aker UT, Heuser RR, King SB, et al. Rapid arterial hemostasis and decreased access site complications after cardiac catheterization and angioplasty: Results of a randomized trial of a novel hemostatic device. Journal of the American College of Cardiology. 1995;25(7):1685-92. ], [2323. Chevalier B, Lancelin B, Koning R, Henry M, Gommeaux A, Pilliere R, et al. Effect of a closure device on complication rates in high-local-risk patients: Results of a randomized multicenter trial. Catheterization and Cardiovascular Interventions. 2003;58(3):285-91. , 2424. Ward SRMD, Casale PMD, Raymond RDO, Kussmaul WG, III, Md, Simpfendorfer CMD. Efficacy and Safety of a Hemostatic Puncture Closure Device With Early Ambulation After Coronary Angiography fn1. American Journal of Cardiology. 1998;81(5):569-72. , 2525. Kapadia SR, Raymond R, Knopf W, Jenkins S, Chapekis A, Ansel G, et al. The 6Fr angio-seal arterial closure device: results from a multimember prospective registry. American Journal of Cardiology. 2001;87(6):789-91. , 2626. Martin JL, Pratsos A, Magargee E, Mayhew K, Pensyl C, Nunn M, et al. A randomized trial comparing compression, perclose proglide™ and Angio-Seal VIP™ for arterial closure following percutaneous coronary intervention: The cap trial. Catheterization and Cardiovascular Interventions. 2008;71(1):1-5. ]. The 6 Fr device was evaluated in a prospective multicentre registry and was proven to be safe providing immediate haemostasis in 91% of patients [2525. Kapadia SR, Raymond R, Knopf W, Jenkins S, Chapekis A, Ansel G, et al. The 6Fr angio-seal arterial closure device: results from a multimember prospective registry. American Journal of Cardiology. 2001;87(6):789-91. ].
The CAP trial compared the Angio-Seal VIP with the Perclose ProGlide and to manual compression with respect to time to haemostasis and ambulation, patient satisfaction, and vascular complications following PCI in 200 patients [2626. Martin JL, Pratsos A, Magargee E, Mayhew K, Pensyl C, Nunn M, et al. A randomized trial comparing compression, perclose proglide™ and Angio-Seal VIP™ for arterial closure following percutaneous coronary intervention: The cap trial. Catheterization and Cardiovascular Interventions. 2008;71(1):1-5. ]. The Angio-Seal was deployed in 100% of patients. The Angio-Seal VIP allowed for earlier haemostasis and ambulation compared with both compression and Perclose ProGlide and was associated with greater patient satisfaction compared with compression (p<0.01).
The Angio-Seal Evolution was evaluated in a multicentre registry involving 10 sites and over 1,000 patients and demonstrated safety and efficacy after routine cardiac catheterisation and PCI [2727. Applegate RJ, Turi Z, Sachdev N, Ahmed A, Szyniszewski A, Foster M, et al. The Angio-Seal Evolution registry: outcomes of a novel automated Angio-Seal vascular closure device. J Invasive Cardiol. 2010;22(9):420-6. ].
Although the effects of Angio-Seal have proved promising, some studies have shown preference for the FemoStop™device [2828. Amin FR, Yousufuddin M, Stables R, Shamim W, Al-Nasser F, Coats AJS, et al. Femoral haemostasis after transcatheter therapeutic intervention: a prospective randomised study of the angio-seal device vs the femostop device. International Journal of Cardiology. 2000;76(2):235-40. ]. The complication and failure rates of Angioseal and the Mynx® were compared in an all-comers population undergoing percutaneous coronary intervention between 2008 and 2014 [2929. Baker NC, Escarcega RO, Lipinski MJ, Magalhaes MA, Koifman E, Kiramijyan S, et al. Active Versus Passive Anchoring Vascular Closure Devices Following Percutaneous Coronary Intervention: A Safety and Efficacy Comparative Analysis. Journal of Interventional Cardiology. 2016;29(1):108-12. ]. 4,074 patients were included, 2,910 receiving the Angioseal and 1,164 receiving Mynx. VCD choice was at the operator’s discretion. Safety was assessed by vascular complications defined as either vascular injury or access-site bleed (haemoglobin drop >3g/dL requiring transfusion, retroperitoneal bleed, or haematoma >5cm, or the composite of both). Efficacy was evaluated by device failure and defined as inability to achieve immediate haemostasis or use of additional haemostatic mechanisms. Outcomes were evaluated at 30-days. The groups were comparable with exceptions as follows: the Angio-Seal group was slightly younger (64±12 vs 65±12, p<0.001), with less peripheral arterial disease (11.3% vs 13.9%, p=0.03) and increased 7Fr sheath use compared with Mynx (59% vs 22%, p<0.001). Safety and efficacy were similar between both groups.
PERCLOSE PROGLIDE SUTURE-MEDIATED CLOSURE (SMC) SYSTEM®
The Perclose® system (Abbott Vascular, Redwood City, CA, USA), introduced in 1994, was the first suture-mediated device to be approved by the Food and Drug Administration. The Perclose Proglide® Suture-Mediated Closure (SMC) system is the latest generation. It offers improvements in the ease of knot delivery and strength and in the non-inflammatory nature of the suture material. It is designed to deliver polyester suture to close femoral artery puncture sites following diagnostic or interventional procedures. The device has one suture and two needles, and a single device can be used to close 5 Fr to 8 Fr access sites.
The Perclose® SMC device is composed of a sheath and a guide. The guide houses the needles and the foot precisely controls the placement of the needles around the puncture site. A marker lumen is contained within the guide, with the intraluminal port of the lumen positioned at the distal end of the needle guide. Proximally, the marker lumen exits from the handle of the device. The marker lumen allows a pathway for back bleeding from the artery to ensure proper device positioning. The device is compatible with a standard 0.038” (or smaller) guidewire.
A knot pusher accessory is included, and is designed to position the tied suture knot to the arteriotomy. The Perclose® Suture Trimmer is also designed to trim the trailing limbs of suture.
The Perclose® system is depicted in Figure 6 and Figure 7.
- Place a 0.038” (or smaller) guidewire through the introducer sheath. Remove the introducer sheath.
- Backload the SMC device over the guidewire until the guidewire skin export port of the sheath is just above the skin line. Remove the guidewire.
- Continue to advance the device until a continuous drop of blood is evident from the marker lumen. Position the device at a 45-degree angle until a continuous drop of blood appears from the marker lumen. Then deploy the foot by pulling the lever back on the top of the handle (marked #1).
- Gently pull the device back to position the foot against the arterial wall. If correct positioning of the foot has been achieved, blood marking will cease. If marking does not stop, gently adjust the angle of the device to stop blood marking.
- While maintaining device position, deploy the needles by pushing on the plunger of the handle (in the direction marked #2).
- Disengage the needles by pulling the plunger back to deploy the suture (in the direction marked #3) and completely remove the plunger and needles from the body of the device.
- Cut the suture from the needles.
- Relax the device, and then return the foot to its original position by pushing the lever (marked #4) on top of the device down to its original position.
- Retract the device until the guidewire port exits the skin line. Remove the two suture limbs (rail suture in blue limb and non-rail in white).
- Insert the guidewire back into the guidewire exit port.
- Withdraw the device from the puncture site while maintain tension on the rail suture (blue),
- Place the rail suture in the snare knot pusher and while maintaining tension on the suture limb with the fingers of one hand, apply forward pressure to the proximal end of the knot pusher with the thumb of the same hand to advance the knot down to the vessel.
- Assess haemostasis and if complete, remove the guidewire while maintaining tension on the rail suture.
- Lock the knot by pulling on the non-rail suture (white).
- Using the right thumb, pull back the thumb knob on the suture trimmer to load both suture limbs.
- Advance suture trimmer down to the vessel and cut the suture by pulling back the red lever with the right index finger.
- Reassess haemostasis by asking the patient to cough or bend leg. If further knot tightening is required, repeat the steps above.
- After successful haemostasis has been achieved, apply an appropriate sterile pressure dressing to the puncture site. Patients can be permitted to sit up in bed immediately after suture and to get up 2–4 hours after suture when no or minimal subcutaneous oozing is present.
PROSTAR® XL PERCUTANEOUS VASCULAR SURGICAL DEVICE
Chapter 3.32
The Prostar® XL Percutaneous Vascular Surgical (Abbott Vascular, Redwood City, CA, USA) device is designed to deliver polyester suture(s) to close femoral artery puncture sites following catheterisation procedures. The Prostar XL is indicated for closure of femoral arterial access in patients who have undergone procedures using 8.5 Fr to 10 Fr sheaths.
The Prostar XL 10 Fr device consists of a J-shaped hydrophilic sheath ( Label A - Figure 8 ) which contains two pairs of sutured needles, a needle guide which precisely controls the needles around the puncture site, and a rotating barrel which receives deployed needles and a marker lumen ( Label B - Figure 8 ). Proximally, the marker lumen exits from the hub of the device and provides a pathway for back bleeding from the femoral artery and ensures proper device positioning. The barrel rotates independently from the central core and is designed to prepare the subcutaneous track via blunt dissection. Barrel rotation is accomplished by depressing the interlocks exiting from the hub. The Prostar XL devices track over a standard 0.038” (or smaller) guidewire.
Other additional accessories are the Perclose® Knot Pusher, which is used to advance the tied knot down to the arteriotomy.
The Perclose Arterial Tamper can be used to augment haemostasis by positioning the knot over the surface of the artery ( Labels A-C - Figure 9 ).
- While the introducer sheath remains in place, a scalpel is used to extend slightly the incision and forceps to dilate the subcutaneous tissue. An 0.038” (or smaller) guidewire is advanced through the introducer sheath. The introducer sheath is removed while applying pressure on the groin to maintain haemostasis. The Prostar XL device is back-loaded over the guidewire until the guidewire exit port is just above the skin line and the guidewire removed.
- The hub is unlocked by depressing the interlocks with thumb and forefinger. Once the hub is unlocked, the hub is rotated while gently advancing the barrel at a 45-degree angle, or less. A steady, continuous drip of blood from the dedicated marker lumen occurs when the Prostar XL device is properly positioned.
- Once in position, the barrel is aligned to lock the hub back in place. While holding the hub with the left hand, the right hand rotates the handle anti-clockwise to unlock the handle. The handle is then pulled away from the hub to deploy the needles. If resistance to rotating the handle is encountered, the device can be backed down, readjusted or exchanged. The handle is pulled until the needle tips emerge at the top of the barrel. While steadily holding the device in position, confirm that all four needles are visible in the hub.
• Suture management
- The sutures are pulled through the arterial wall and removed from the back end of the barrel. One green 3-0 braided polyester suture is deployed at the 10 and 4 o’clock positions and one white 3-0 polyester braided suture is deployed at the 2 and 8 o’clock positions. The needles are removed from the hub using a haemostat. The needles are cut off from the sutures and the sutures re-tagged with haemostats to prevent them from being pulled out of the artery.
- The slack is removed in the suture(s) by pulling the suture ends to evenly matched lengths and by tensioning until resistance is felt.
- The Prostar XL device is gently withdrawn until the guidewire port exits the skin line. The guidewire is reinserted through the side port to regain access to the artery and to allow for the removal of the device.
- At the end of the procedure, the sutures should be cleaned with saline. The sheath is removed and either a slipknot or a modified fisherman’s knot is tied while a second operator performs manual compression proximal to the site of the puncture. The white knot is tied first and the Knot Pusher used to cinch the knot down onto the artery. After this, the guidewire is removed and the green suture is tied. With the Knot Pusher in place, tighten the knot by gently pulling on the white non-rail suture. Once haemostasis is achieved the sutures are trimmed below the skin and manual compression performed for approximately 5 minutes.
The “Closer” devices can be used with several different sheath sizes. “Perclose™” can be performed prior to sheath insertion when using very large sheaths, allowing the operator to create the ‘’purse-string’’ before actually dilating the arteriotomy. This is especially useful in percutaneous aortic valve implantations and closures of abdominal aortic aneurysms, where sheath sizes currently approach 18 Fr.
Trial data for the closer devices
The major trials for the Perclose® device were the STAND I and STAND II trials. The STAND I trial (a non-randomised registry) evaluated the device in 200 patients undergoing diagnostic procedures with successful haemostasis in 99% of patients in a median time of 13 minutes and with a major complication rate of 1% [1717. Baim DS, Knopf WD, Hinohara T, Schwarten DE, Schatz RA, Pinkerton CA, et al. Suture-mediated closure of the femoral access site after cardiac catheterization: results of the suture to ambulate aNd discharge (STAND I and STAND ii) trials. American Journal of Cardiology. 2000;85(7):864-9. ].
The STAND II trial was the pivotal randomised controlled multicentre trial [1717. Baim DS, Knopf WD, Hinohara T, Schwarten DE, Schatz RA, Pinkerton CA, et al. Suture-mediated closure of the femoral access site after cardiac catheterization: results of the suture to ambulate aNd discharge (STAND I and STAND ii) trials. American Journal of Cardiology. 2000;85(7):864-9. ]. 515 patients undergoing diagnostic or interventional procedures were randomised to the use of the 8 Fr or the 10 Fr Prostar-Plus device versus manual compression. Successful haemostasis was achieved in 97.6% of patients compared with 98.9% by compression (p=ns). In the diagnostic cases, the time to discharge was significantly reduced from 8.3 hours in the compression arm to 4.4 hours in the Prostar arm. In the interventional cases, time to discharge remained unaltered. There was a slight trend towards increased complications with the suture-based device but this was not statistically significant. The STAND II trial proved the safety and efficacy of a suture-based concept for arteriotomy closure.
The safety and efficacy of the Perclose® has been supported by the results of other randomised studies [1717. Baim DS, Knopf WD, Hinohara T, Schwarten DE, Schatz RA, Pinkerton CA, et al. Suture-mediated closure of the femoral access site after cardiac catheterization: results of the suture to ambulate aNd discharge (STAND I and STAND ii) trials. American Journal of Cardiology. 2000;85(7):864-9. ], [3030. Gerckens U, Cattelaens N, Lampe E-G, Grube E. Management of arterial puncture site after catheterization procedures: evaluating a suture-mediated closure device. American Journal of Cardiology. 1999;83(12):1658-63. , 3131. Carere RG, Webb JG, Buller CEH, Wilson M, Rahman T, Spinelli J, et al. Suture closure of femoral arterial puncture sites after coronary angioplasty followed by same-day discharge. American Heart Journal. 2000;139(1):52-8. , 3232. Noguchi T, Miyazaki S, Yasuda S, Baba T, Sumida H, Morii I, et al. A randomised controlled trial of Prostar Plus for haemostasis in patients after coronary angioplasty. Eur J Vasc Endovasc Surg. 2000;19(5):451-5. , 3333. Wetter DR, Rickli H, von Smekal A, Amann FW. Early sheath removal after coronary artery interventions with use of a suture-mediated closure device: clinical outcome and results of Doppler US evaluation. J Vasc Interv Radiol. 2000;11(8):1033-7. , 3434. Rickli H, Unterweger M, Sütsch G, Brunner-La Rocca HP, Sagmeister M, Ammann P, et al. Comparison of costs and safety of a suture-mediated closure device with conventional manual compression after coronary artery interventions. Catheterization and Cardiovascular Interventions. 2002;57(3):297-302. , 3535. Nasu K, Tsuchikane E, Sumitsuji S. Clinical effectiveness of the Prostar XL suture-mediated percutaneous vascular closure device following PCI: results of the Perclose AcceleRated Ambulation and DISchargE (PARADISE) Trial. J Invasive Cardiol. 2003;15(5):251-6. ].
The newest generation device was evaluated in 72 procedures with a success rate of 98.6% without any notable complications [3636. Shimshak T. Vascular access site management in high risk patients. Endovascular Today. 2003;2:48-50. ] . Immediate ambulation with this device has been shown to be feasible and safe [3737. Dorogy ME, Class S, Sprague K, Breall J, Kalaria VG. Safety of immediate ambulation after therapeutic intervention. 2003. 149L-L p ].
A useful feature of the Perclose® system is the ability for "pre-closure" [3838. Feldman T. Femoral arterial preclosure: finishing a procedure before it begins. Catheter Cardiovasc Interv. 53 United States 2001 p 448. ]. The sutures are deployed at the beginning of the procedure using a smaller sheath, which is then exchanged for the Perclose® device. A larger sheath is then placed and the sutures tied after completion of the procedure. This method has been used successfully with 7-8 Fr sheaths as well as with larger [1010. Koreny M, Riedmüller E, Nikfardjam M, Siostrzonek P, Müllner M. Arterial puncture closing devices compared with standard manual compression after cardiac catheterization: Systematic review and meta-analysis. JAMA. 2004;291(3):350-7. , 1111. Piper WD, Malenka DJ, Ryan TJ, Shubrooks SJ, O’Connor GT, Robb JF, et al. Predicting vascular complications in percutaneous coronary interventions. American Heart Journal. 2003;145(6):1022-9. , 1212. Tavris DR, Gallauresi BA, Lin B, Rich SE, Shaw RE, Weintraub WS, et al. Risk of local adverse events following cardiac catheterization by hemostasis device use and gender. J Invasive Cardiol. 2004;16(9):459-64. , 1313. Tavris D, Gross T, Gallauresi B, Kessler L. Arteriotomy closure devices—the FDA perspective - Editorials published in the Journal of the American College of Cardiologyreflect the views of the authors and do not necessarily represent the views of JACCor the American College of Cardiology. Journal of the American College of Cardiology. 2001;38(3):642-4. , 1414. Dauerman HL, Applegate RJ, Cohen DJ. Vascular Closure Devices: The Second Decade. Journal of the American College of Cardiology. 2007;50(17):1617-26. , 1515. Nikolsky E, Mehran R, Halkin A, Aymong ED, Mintz GS, Lasic Z, et al. Vascular complications associated with arteriotomy closure devices in patients undergoing percutaneous coronary procedures: A meta-analysis. Journal of the American College of Cardiology. 2004;44(6):1200-9. , 1616. Vaitkus PT. A meta-analysis of percutaneous vascular closure devices after diagnostic catheterization and percutaneous coronary intervention. J Invasive Cardiol. 2004;16(5):243-6. ] Fr sheaths [3939. Bhatt DL, Raymond RE, Feldman T, Braden GA, Murphy B, Strumpf R, et al. Successful "pre-closure" of 7Fr and 8Fr femoral arteriotomies with a 6Fr suture-based device (the Multicenter Interventional Closer Registry). American Journal of Cardiology. 2002;89(6):777-9. , 4040. Solomon LW, Fusman B, Jolly N, Kim A, Feldman T. Percutaneous suture closure for management of large French size arterial puncture in aortic valvuloplasty. J Invasive Cardiol. 2001;13(8):592-6. , 4141. Torsello GB, Kasprzak B, Klenk E, Tessarek J, Osada N, Torsello GF. Endovascular suture versus cutdown for endovascular aneurysm repair: a prospective randomized pilot study1. Journal of Vascular Surgery. 2003;38(1):78-82. ] used for percutaneous transaortic valve implantations and endoluminal repair of abdominal aortic aneurysms.
Reports have shown safety when closing up to 22 Fr femoral arteriotomies [4242. Krajcer Z, Howell M. A novel technique using the percutaneous vascular surgery device to close the 22 French femoral artery entry site used for percutaneous abdominal aortic aneurysm exclusion. Catheter Cardiovasc Interv. 2000;50(3):356-60. ] and brachial access sites [4343. Kim A, Fusman B, Jolly N, Feldman TED. Percutaneous Suture Closure for Brachial Artery Puncture. Journal of Interventional Cardiology. 2002;15(4):277-80. ]. The device can be used several times within days and the device can be used multiple times within the same femoral artery [4444. Razminia M, Molnar J, Kunjummen B. Repeated arteriotomy repair using suture-mediated closure following transfemoral diagnostic and therapeutic intervention. Am J Cardiol. 2002;24:168H. ]. The CONTROL study (ClOsure device iN TRansfemoral aOrtic valve implantation) multi-centre study included 3138 consecutive percutaneous transfemoral TAVI patients, categorized according to vascular closure strategy: Prostar XL vs Perclose ProGlide-based vascular closure strategy [4545. Barbash IM, Barbanti M, Webb J, Molina-Martin De Nicolas J, Abramowitz Y, Latib A, et al. Comparison of vascular closure devices for access site closure after transfemoral aortic valve implantation. European Heart Journal. 2015;36(47):3370-9. ]. Propensity score matching identified 944 well-matched patients. Composite primary endpoint of major vascular complications or in-hospital mortality occurred more frequently in the Prostar group when compared with the ProGlide group (9.5% vs 5.1%, p=0.016) and was driven by higher rates of major vascular complication (7.4% vs 1.9%, p<0.001) in the Prostar group. In hospital mortality was similar between the groups. Prostar use was associated with higher rates of major bleeding (16.7% vs 3.2%, p<0.001), acute kidney injury (17.6% vs 4.4%, p<0.001) and with longer hospital stay (median 6 vs 5 days, p=0.007).
A recent retrospective study evaluated major and minor bleeding complications as defined by VARC following balloon aortic valvuloplasty in 930 patients [4646. Dall’Ara G, Santarelli A, Marzocchi A, Bacchi Reggiani ML, Sabattini MR, Moretti C, et al. Vascular complications after balloon aortic valvuloplasty in recent years: Incidence and comparison of two hemostatic devices. Catheterization and Cardiovascular Interventions. 2018;91(6):E49-E55. ]. Vascular closure was performed with Angioseal in 69% and with Perclose in 31% of cases. The size of sheath was 9Fr in the majority of cases (84.1%). The Angioseal group had a higher rate of small haematomas (6.9% v 3.5%, p=0.042) but fewer blood transfusions (3.5% v 6.6%, p=0.034). There were no significant differences in in-hospital MACCE and 30 day survival between both devices.
FEMOSEAL™
The FemoSeal™ (Terumo Medical Corporation, NJ, USA) is an invasive closure device designed to induce quick and effective haemostasis ( Figure 10 and Figure 11 ). This mechanical device delivers haemostasis without collagen or other sealing agents. FemoSeal™ consists of two resorbable polymer discs, an inner seal deployed intra-arterially and an outer locking disc deployed on the outer wall of the artery. Haemostasis is achieved by sealing the arteriotomy between the two discs, which are held together by a resorbable multifilament complete with friction lock. Haemostasis is achieved once the inner lock is deployed inside the artery. The device automatically signals when you should deploy the outer locking disc, thereby sealing the arteriotomy.
Trial data for FemoSeal™
The FemoSeal™ vascular closure device was used in the treatment of 1058 patients in the SCAAR registry and was shown to be safe with a bleeding rate of 0.2%. [4747. James S, Lagerqvist B. SCAAR registry annual report 2007. 2007. ]
The CLOSE-UP study was a randomised controlled trial of 1001 patients enrolled in Denmark comparing the Femoseal device with manual compression [4848. Holm NR, Sindberg B, Schou M, Maeng M, Kaltoft AK, Bottcher M, et al. Randomised comparison of manual compression and FemoSeal™ vascular closure device for closure after femoral artery access coronary angiography: the CLOSure dEvices Used in everyday Practice (CLOSE-UP) study. EuroIntervention. 2014;10(2):183-90. ]. The primary endpoint of the study of large groin haematomas (defined as >5 cm) was statistically lower in the Femoseal group (2.2% v 6.7%, p=0.002). Combined endpoint of 14-day adverse vascular events was similar in both groups.
Alonzo et al compared the MACCE and TIMI bleeding rates between transfemoral route sealed with Femoseal and transradial access in a cohort of patients undergoing primary PCI [4949. Alonzo A, Rigattieri S, Giovannelli F, Di Russo C, Sciahbasi A, Berni A, et al. Transfemoral approach with systematic use of FemoSeal™ closure device compared to transradial approach in primary angioplasty. Catheterization and Cardiovascular Interventions. 2016;87(5):849-54. ]. This was a retrospective and propensity-matched cohort of 777 patients with 511 in the transfemoral and 266 in the transdradial groups. The TIMI bleeding rate in the Femoseal group was 6.6% and statistically higher than that of the radial group (1.3%).
EXOSEAL®
The ExoSeal® (Cordis, Johnson & Johnson, Warren, NJ, USA) VCD is a bioabsorbable device designed for the sealing of femoral artery puncture sites in patients who have undergone diagnostic or interventional procedures using a standard 6 Fr introducer sheath and is indicated for closure of 5F, 6F and 7F arteriotomies. The device achieves haemostasis by means of a visually-guided deployment mechanism which delivers a bioabsorbable polyglycolic acid "plug" atop the femoral artery, anchored by the neurovascular bundle sheath. The plug, which is entirely extravascular, is subsequently hydrolysed into CO2 and H2O via the Krebs cycle over a 3-month period. The device is not suitable for use through sheaths that are longer than 12cm and should not be used in vessel of diameters <5mm.
- The system is introduced directly into the sheath until the black marker is reached.
- The system is clipped into the sheath. This is followed by the appearance of a pulsatile flow of blood through the Bleed-Back System.
- The system is slowly withdrawn while watching the two black and white markers on the upper surface of the system.
- Initially the flow of blood stops then the black and white marker turns entirely black.
- The system is released by pressing the green button.
- A non-occlusive dressing is applied for 2-3 minutes.
Trial data for the ExoSeal®
The pivotal trial of the ExoSeal® vascular closure device was the ECLIPSE trial. This was a 2:1, randomised non-blinded multicentre US trial involving 401 patients comparing the ExoSeal with manual compression. The procedural success rate was 91% with no statistical difference between the groups. Time to haemostasis (4.4 vs. 20.1 minutes) and time to ambulation (2.54 vs. 6.24 hours) were both statistically in favour of the ExoSeal device. There were no major vascular complications [5050. Wong SC, Bachinsky W, Cambier P, Stoler R, Aji J, Rogers JH, et al. A Randomized Comparison of a Novel Bioabsorbable Vascular Closure Device Versus Manual Compression in the Achievement of Hemostasis After Percutaneous Femoral Procedures: The ECLIPSE (Ensure’s Vascular Closure Device Speeds Hemostasis Trial). JACC: Cardiovascular Interventions. 2009;2(8):785-93. ].
A 1:1 randomised trial comparing the Proglide with the Exoseal enrolled 100 patients undergoing PCI and endovascular peripheral procedures with end points of immediate total haemostasis and incidence of vascular complication. Immediate haemostasis was more frequent and vascular complication less frequent with the Exoseal device but this did not reach statistical significance [5151. Hattab M, Hakim M, Carreira VB, Elhadad S. TCT-393 A randomized trial comparing two vascular closure devices: PROGLIDE and the novel EXOSEAL after percutaneous femoral procedures. Journal of the American College of Cardiology. 2012;60(17 Supplement):B112. ].
In contrast, other study found a statistically higher incidence of vascular complications with this device when compared to either Angioseal or Perclose (3.6% v 1.2%) [5252. Kara K, Mahabadi AA, Rothe H, Müller P, Krüger J, Neubauer H, et al. Safety and Effectiveness of a Novel Vascular Closure Device: A Prospective Study of the ExoSeal Compared to the Angio-Seal and ProGlide. Journal of Endovascular Therapy. 2014;21(6):822-8. ]. Logistic regression analysis showed that the use of the Exoseal device was associated with a three fold odds of complications. Repeat angiography performed a month after deployment of the Exoseal device shows angiographic irregularities in nearly 7% of patients treated [5353. Grandhi R, Zhang X, Panczykowski D, Choi P, Hunnicutt CT, Jadhav AP, et al. Incidence of delayed angiographic femoral artery complications using the EXOSEAL vascular closure device. Interventional neuroradiology : journal of peritherapeutic neuroradiology, surgical procedures and related neurosciences. 2015;21(3):401-6. ].
MYNX® VASCULAR CLOSURE DEVICE
The Mynx® Vascular Closure Device (Cardinal Health, Dublin, OH, USA) is an extravascular vascular closure device (VCD) whose deployment system is designed to minimise the discomfort commonly associated with closing the femoral artery following a catheterisation procedure. The Mynx® received FDA Premarket Approval (PMA) in May 2007.
The Mynx® uses a soft, sponge-like sealant material to close the arteriotomy ( Figure 13 ) The sealant works by rapidly absorbing blood and fluids around the puncture site, swelling in size and covering the hole. This seals the hole and stops the bleeding.
The sealant material consists of polyethylene glycol (PEG), a water-soluble, non-thrombogenic, conformable, bio-inert polymer which is fully resorbed by the body within 30 days. The Mynx® device uses an extravascular method of deployment. It avoids cinching or tugging of the artery by placing the Mynx® sealant on the outside of the artery. The Mynx® uses the existing 5, 6 or 7 Fr procedural sheath, eliminating the need for a sheath exchange. This avoids potential tissue tract expansion, which can cause tissue trauma and oozing, often resulting in longer bed rest for the patient and potentially delaying ambulation and discharge.
As the Mynx® sealant is placed on the outside of the artery, it is exposed to blood and subcutaneous fluids. The sealant rapidly expands, covering the hole in the artery and conforming to the tissue tract, producing haemostasis. This extravascular placement of the sealant avoids leaving behind an intravascular component, which can compromise blood flow and, in rare circumstances, embolise, requiring surgical repair. The Mynx® sealant avoids initiating platelet activation or an abnormal inflammatory response, and does not generate fibrous scar tissue. In addition, the porous characteristics of the sealant create a platform for natural clot formation and facilitate tissue healing.
The Mynx® VCD includes a balloon catheter with integrated sealant and a 10 ml lock syringe.
- The Mynx® device is inserted into the existing femoral sheath. A small semi-compliant balloon is inflated to achieve haemostasis.
- The sealant is delivered through the introducer sheath, exposing it to blood and subcutaneous fluid, producing a durable haemostasis
- The balloon is deflated and the device removed. The sealant is now on the surface of the arteriotomy
- Within 30 days, the sealant has dissolved leaving nothing behind.
The current commercially available devices are the MynxAce ( Figure 15 ) and MynxGrip ( Figure 16 ) vascular closure devices.
The MynxAce device combines the deployment system and extravascular sealant as described above. MynxGrip adds the proprietary Grip Technology to the distal end of the original Mynx Sealant. The result is a sealant that adheres to and seals the arteriotomy while expanding to fill the tissue tract.
Trial data for the MYNX®
This device was studied in a prospective multicentred single arm clinical investigation at 5 European centres. 190 patients were included with 50% undergoing a diagnostic procedure and 50% intervention with sheath sizes 5-7 Fr. The device was found to have a major complication rate of 0.5% in this low to intermediate risk group and no device precipitated complications associated with clinical sequelae were reported [5454. Scheinert D, Sievert H, Turco MA, Schmidt A, Hauptmann KE, Mueller R, et al. The safety and efficacy of an extravascular, water-soluble sealant for vascular closure: Initial clinical results for Mynx™. Catheterization and Cardiovascular Interventions. 2007;70(5):627-33. ].
However other registries have found a significant incidence of pseudoaneurysms [5555. Fields JD, Liu KC, Lee DS, Gonda SJ, Dogan A, Gultekin SH, et al. Femoral Artery Complications Associated with the Mynx Closure Device. American Journal of Neuroradiology. 2010;31(9):1737. ], vascular complications [5656. Kumar A, Matheny ME, Ho KKL, Yeh RW, Piemonte TC, Waldman H, et al. The data extraction and longitudinal trend analysis network study of distributed automated postmarket cardiovascular device safety surveillance. Circulation Cardiovascular quality and outcomes. 2015;8(1):38-46. , 5757. Tavris DR, Wang Y, Jacobs S, Gallauresi B, Curtis J, Messenger J, et al. Bleeding and vascular complications at the femoral access site following percutaneous coronary intervention (PCI): an evaluation of hemostasis strategies. J Invasive Cardiol. 2012;24(7):328-34. ] and device failure [5858. Azmoon S, Pucillo AL, Aronow WS, Ebrahimi R, Vozzolo J, Rajdev A, et al. Vascular complications after percutaneous coronary intervention following hemostasis with the Mynx vascular closure device versus the AngioSeal vascular closure device. J Invasive Cardiol. 2010;22(4):175-8. ]. These concerns led to a large scale analysis of the NCDR Cath PCI registry to assess the safety of this device [5959. Resnic FS, Majithia A, Marinac-Dabic D, Robbins S, Ssemaganda H, Hewitt K, et al. Registry-Based Prospective, Active Surveillance of Medical-Device Safety. N Engl J Med. 2017;376(6):526-35. Epub 2017/01/26. ]. The report showed that the Mynx device was associated with a significantly greater risk of vascular complications (1.2% v 0.8%, p< 0.001), access site re-bleeding (0.4% v 0.3%, p=0.001) and need for transfusion (1.8% v 1.5%, p<0.001) than alternative VCDs.
FISH™ DEVICE
The Femoral Introducer Sheath and Haemostasis (FISH™) Device (Morris Innovative Research, Bloomington, IN, USA) is used to stop bleeding at a puncture site following 5, 6, or 8 Fr cardiac catheterisation procedures. It is a patching device using porcine biomaterial labelled as small intestinal submucosa (SIS) attached to the introducer sheath. Theoretically, the presence of the patch at the arterial entry site during the procedure favours early healing. Following catheterisation, a release wire is pulled detaching the patch from the sheath. The vessel wall is remodelled within 30 days. The FISH™ device also allows for the option of delayed sheath removal in the recovery ward and discontinuation of anticoagulants. The FISH™ device can be used as a combined procedural sheath and closure device, or placed after the procedure and used as a closure device only ( Figure 25_2016.jpg" data-toggle="modal" data-target="#popup-media" class="media-link" data-media_id="3284" data-folder="pcr-textbook" data-chapterid="68"> Figure 17 ).
Trial data for FISH™
The FISH™ closure device was studied in a randomised controlled multicentre clinical trial with almost 300 patients. The study included patients undergoing diagnostic or therapeutic coronary or peripheral procedures performed via the common femoral artery. The procedures were done with an 8 Fr or smaller sheath size. Patients were randomised in a 2:1 fashion to the FISH device or manual compression with primary endpoints of haemostasis and time to ambulation. The results showed a significantly shorter time to haemostasis and mean time to ambulation with the FISH device. There were no significant differences in adverse vascular events. However, it should be noted that 27 patients converted from the FISH device to manual compression [6060. Bavry AA, Raymond Re Fau - Bhatt DL, Bhatt Dl Fau - Chambers CE, Chambers Ce Fau - DeNardo AJ, DeNardo Aj Fau - Hermiller JB, Hermiller Jb Fau - Myers PR, et al. Efficacy of a novel procedure sheath and closure device during diagnostic catheterization: the multicenter randomized clinical trial of the FISH device. (1557-2501 (Electronic)). ].
There have been no studies comparing the FISH device to the market leaders to date.
STARCLOSE®
The StarClose® (Abbott Vascular, Redwood City, CA, USA) is a clip-mediated closure device. It was approved by the Food and Drug Administration in 2005 but has been successfully used in Europe since 2004. In January 2007, StarClose® was approved for interventional use. The StarClose® introduces a small, circumferential, flexible clip, which mechanically binds the surface of the femoral artery together ( Figure 18 ). The clip is made of nitinol, a nickel titanium alloy whose superelastic properties allow it to return to its original shape once released from the device. Its use involves a 4-step or click process: (1) replacement of the procedural sheath with the StarClose® sheath, (2) deployment of the vessel locator, (3) delivery of the clip, and (4) deployment of the clip, achieving haemostasis. Because the clip is on the outside of the artery, nothing is left behind on the inside to cause potential blockages later.
- To deploy the StarClose®, the arterial sheath present at the end of the procedure is removed and the groin re-prepared. A 5-7 mm incision is made at the puncture site and the arteriotomy site enlarged ( Figure 19AB )
- A dedicated 6 Fr StarClose® Exchange Sheath is introduced over the wire and the carrier tube containing the clip is then connected to the sheath ( Figure 19CD ) audible "click" will be heard.
- The vessel locator button on the end of the device is pushed. A "muted click" will be heard. The device is retracted 1-2 cm and the thumb advancer advanced 1-2 cm in order to split the sheath above the skin ( Figure 19EF ).
- The device is maintained at the angle of the tissue track and pulled back until tactile resistance is felt. The thumb advancer is advanced until it meets the finger loop and the sheath split ( Figure 19GH ). An audible "click" will be heard.
- The device is raised to 60-75 degrees and the clip deployed ( Figure 19IJ ). An audible "click" will be heard.
Trial data for the StarClose®
The pivotal trial demonstrating the safety and efficacy of the StarClose® Vascular Closure System was the Clip closure In Percutaneous procedures (CLIP) study [6161. Jaff MR, Hadley G, Hermiller JB, Simonton C, Hinohara T, Cannon L, et al. The safety and efficacy of the StarClose® vascular closure system: The ultrasound substudy of the CLIP study. Catheterization and Cardiovascular Interventions. 2006;68(5):684-9. ]. This multicentre study enrolled 596 patients in 17 US centres who were undergoing diagnostic and interventional procedures. Patients were randomised in a 2:1 fashion to receive the StarClose VCD or standard compression. 208 patients underwent diagnostic angiography only. In this subgroup, there was no difference in the rates of major and minor vascular complications between the StarClose VCD and manual compression. The device was deployed successfully in 94% of patients and successfully reduced the mean time to haemostasis (15.5 vs. 1.5 minutes, p<0.001) and time to ambulation (268 to 163 minutes, p<0.001) [88. Sherev DA, Shaw RE, Brent BN. Angiographic predictors of femoral access site complications: Implication for planned percutaneous coronary intervention. Catheterization and Cardiovascular Interventions. 2005;65(2):196-202. ]. These findings were confirmed in the interventional subset with no differences in the rates of major complications and a trend towards a lower rate of minor complications in the StarClose group [2020. Hermiller JB, Simonton C, Hinohara T, Lee D, Cannon L, Mooney M, et al. The StarClose® vascular closure system: Interventional results from the CLIP study. Catheterization and Cardiovascular Interventions. 2006;68(5):677-83. ].
A smaller prospective study enrolled 71 patients undergoing diagnostic cardiac or peripheral angiography via the femoral approach in 11 US centres. The 30-day outcomes showed 0% of major complications and 3% of minor complications consistent with the findings of the CLIP study [6262. Burke MN, Hermiller J, Jaff MR. StarClose® vascular closure system (VCS) is safe and effective in patients who ambulate early following successful femoral artery access closure—results from the RISE clinical trial. Catheterization and Cardiovascular Interventions. 2012;80(1):45-52. ].
A recent randomised controlled trial comparing StarClose and Proglide for femoral haemostasis in patients found a statistically significantly higher rate of vascular complications with StarClose (5.6% v 2.2%) [6363. Klein-Wiele O, Baliota M, Kara K, Käunicke M, Schäfer H, Garbrecht M, et al. Safety and efficacy of clip-based vs suture mediated vascular closure for femoral access hemostasis: A prospective randomized single center study comparing the StarClose and the ProGlide device. Catheterization and Cardiovascular Interventions. 2018;91(3):402-7. ].
CELT-ACD®
The Celt ACD® (Vasorum Ltd, Trinitas House, Dublin, Ireland) achieves closure by applying a biocompatible implant to both the inside and the outside of the puncture site in the femoral artery (which is deep to the skin in the groin area) at the level of the arterial wall. It is available in three sizes to fit 5,6 and 7Fr sheaths. This device does not require exchange of the femoral sheath before deployment ( Figure 20 ).
Trial data for CELT-ACD®
The CELT ACD trial compared the CELT-ACD against manual compression for femoral site haemostasis in 207 patients undergoing coronary procedures using a 6Fr sheath [6464. Wong SC, Laule M, Turi Z, Sanad W, Crowley J, Degen H, et al. A multicenter randomized trial comparing the effectiveness and safety of a novel vascular closure device to manual compression in anticoagulated patients undergoing percutaneous transfemoral procedures: The CELT ACD trial. Catheterization and Cardiovascular Interventions. 2017;90(5):756-65. ]. The primary endpoint was time to haemostasis while the secondary endpoint was access site complication rate at 30 days. The time to haemostasis was significantly lower in the device group (0 v 8 mins, p< 0.0001) while the 30 day vascular complication rates were similar in both groups.
CARDIVA CATALYST®
The Cardiva Catalyst® (Cardiva Medical, Mountain View, CA, USA) is a deployable nitinol disc sheathed in a biocompatible membrane, intended to promote haemostasis at an arteriotomy site as an adjunct to manual compression ( Figure 21A ). Cardiva Catalyst® is indicated for use in patients undergoing diagnostic and/or interventional femoral artery catheterisation procedures using up to 7 Fr sheaths.
The Cardiva Catalyst® consists of a sterile disposable Catalyst® wire and a sterile disposable Catalyst Clip. The wire is available in two versions – Cardiva Catalyst® II and III. The Catalyst II wire contains two agents (kaolin and chitosan) which, when exposed to the tissue tract, accelerate haemostasis by stimulating coagulation, platelet adhesion, and platelet aggregation. The Catalyst III wire has an additional protamine coating which, when exposed to a tissue tract, provides rapid haemostasis in patients anticoagulated with heparin. Catalyst III is indicated for heparinised patients undergoing femoral percutaneous interventions.
The Cardiva Catalyst® still needs assisted manual compression once the disc is collapsed and retracted out of the vessel.
Procedure ( Figure 21B )
- After completion of the catheterisation procedure, the Catalyst Wire is inserted into the artery through the existing introducer sheath.
- The distal tip of the Catalyst wire is deployed, which opens the bi-convex, low-profile Catalyst Disc within the lumen of the femoral artery distal to the introducer sheath tip.
- After removing the introducer sheath over the Catalyst Wire, gentle upward tension is applied to the Catalyst Wire to conform the Catalyst Disc to the contours of the vessel securing it against the intima, blocking the arteriotomy. Tension is maintained by applying the Catalyst Clip externally to the Catalyst II Wire at the puncture site. Tension between the Catalyst Disc and the Catalyst Clip creates a site-specific compression of the arteriotomy and establishes temporary haemostasis. During dwell, natural recoil of smooth muscle in the vessel wall occurs at the arteriotomy site. A biocompatible coating on the Catalyst Wire aids the body's natural haemostatic process and promotes ease of removal.
- Following appropriate dwell time, the Catalyst Disc is collapsed and the Cardiva Catalyst is completely removed from the artery. No part of the device is left behind.
- Final haemostasis of the vessel puncture site occurs with application of manual or mechanical compression after removing the Cardiva Catalyst.
Trial data for Cardiva Catalyst®
This device has been studied in 96 patients following diagnostic cardiac catheterisation, including 25 patients (26%) with contraindications to conventional closure devices. Femoral angiography was performed prior to deployment of the Cardiva closure device. Patients were ambulated at 1 hour after haemostasis was achieved. Successful deployment was achieved in 99% of patients with no major complications and a minor complication rate of 5% [6565. Doyle BJ, Godfrey MJ, Lennon RJ, Ryan JL, Bresnahan JF, Rihal CS, et al. Initial experience with the cardiva Boomerang™ vascular closure device in diagnostic catheterization. Catheterization and Cardiovascular Interventions. 2007;69(2):203-8. ].
The Boomerang trial was a prospective, multicentre, randomized, controlled trial comparing the use of the Boomerang wire (now Cardiva Catalyst®) in conjunction with manual compression versus manual compression alone to achieve haemostasis in diagnostic and interventional procedures via the femoral approach [6666. Goswami NJ, Smalling RG, Sinha S, Gammon RS, Ramaiah VG. Comparison of the boomerang wire vascular access management system versus manual compression alone during percutaneous diagnostic and interventional cardiovascular procedures: The boomerang™ wire vascular access management trial II. Catheterization and Cardiovascular Interventions. 2016;87(1):75-81. ]. Subjects were randomized 3:1 to Boomerang versus manual compression. No minor or major device-related adverse events were reported. Non-device related complications were 3 (0.9%) in the Boomerang arm (n=327) and 1 (0.8%) in the manual compression arm (n=123). Mean time to haemostasis and time to ambulation were significantly reduced in the Boomerang arm for both diagnostic and interventional procedures (p<0.0001).
VASCADE® VASCULAR CLOSURE SYSTEM
The Vascade® Vascular closure system is the third generation of extravascular closure device from Cardiva Medical ( Figure 22 ). Indicated for femoral arterial access site closure in patients who have undergone diagnostic or interventional endovascular procedures using a 5F, 6F or 7F procedural sheath, it reduces the time to haemostasis and ambulation.
Vascade® combines both mechanical and biochemical methods of haemostasis for closure of the femoral arterial access site. The main difference with the third generation device is the addition of a protamine sulphate component on the coating applied to the extravascular anchor element in addition to the two other proprietary coatings in the Catalyst® II. The main advantages of the device are a lack of biologic material or permanent implant, as well as the ability to apply it to a wide variety of anatomical conditions [6565. Doyle BJ, Godfrey MJ, Lennon RJ, Ryan JL, Bresnahan JF, Rihal CS, et al. Initial experience with the cardiva Boomerang™ vascular closure device in diagnostic catheterization. Catheterization and Cardiovascular Interventions. 2007;69(2):203-8. ].
Trial data for Vascade® Vascular Closure system
The safety and effectiveness of Vascade® Vascular Closure System (VCS) was evaluated in 420 patients across 21 clinical sites up to 30 days post diagnostic or interventional procedures in the RESPECT trial with patients randomised 2:1 to VASCADE versus manual compression [6767. Hermiller JB, Leimbach W, Gammon R, Karas SP, Whitbourn RJ, Wong SC, et al. A prospective, randomized, pivotal trial of a novel extravascular collagen-based closure device compared to manual compression in diagnostic and interventional patients. J Invasive Cardiol. 2015;27(3):129-36. ]. There was a high proportion of bivalirudin use in both groups being slightly higher in the Vascade group. (77% vs 69%). The clinical data show that patients treated with the Vascade VCS had a lower mean time to haemostasis and ambulation than the corresponding times for those patients treated with manual compression, and that the differences in these times are statistically and clinically significant. In addition, diagnostic patients treated with the Vascade VCS had a lower mean time to discharge eligibility than the corresponding times for those patients treated with manual compression, and the difference in these times is statistically and clinically significant. The primary safety endpoint was the combined rate of major complications within 30 ± 7 days following the catheterization procedure. The secondary safety endpoint was the combined rate of minor complications within 30 ± 7 days following the catheterization procedure. There were no major complications in the pivotal study in either arm. Additionally, for the total patients the rate of overall minor complications was statistically significantly less in the Vascade treatment arm as compared to manual compression.
AXERA®
The Axera® device (Arstasis, Redwood City, CA) is the second-generation device from this company and is a single use system for the provision for vascular access and also to promote haemostasis of the vascular site as an adjunct to manual compression ( Figure 23A ). The system employs a strategy of preclosure and utilises a novel strategy whereby a controlled preprocedure arteriotomy results in an overlap of arterial tissue after sheath removal that is reinforced by hydrostatic arterial pressure that creates closure. This approach leaves no foreign material behind. The Axera device is approved for used in diagnostic cases with 5F or 6F sheaths.
The Arstasis™ family of devices uses a traditional femoral entry approach. Following the needle stick and wire insertion, the Axera® is deployed in 3 steps ( Figure 23B, Figure 23C and Figure 23D ).
- Step 1 – Introduce Access Device
The 0.035” Latchwire is introduced through the access needle and the needle then removed. The Latchwire is attached to the distal top of the Axera® device. The device is then advanced into the artery until blood is observed through the marker port.
- Step 2 – Create Self-sealing Arteriotomy
The heel actuator is slid back to deploy the heel inside the artery. The Axera® is then retracted at 55-60° until resistance is felt and the blood mark stops or diminishes. The plunger is depressed to advance the micropuncture needle and create a self-sealing arteriotomy. The 2nd mark should be observed through the marker port.
The guidewire is introduced through the plunger port and the micropuncture needle. The plunger is retracted to withdraw the needle and the heel released. The device is removed. The sheath, dilator and dilator adaptor assembly is then advanced. The sheath may be removed when clinically indicated. Brief manual compression, above the puncture site, is then applied until haemostasis is achieved.
Trial data for the Axera®
The RECITAL study (68) was a prospective multi centred study which enrolled 351 patients to assess device and procedural success of key post-catheterization recovery measures that influence the safety and speed toward discharging those patients from the hospital. Key results of the RECITAL study showed that device deployment was successful in 97% of RECITAL patients and femoral bleeding stopped after only 3 minutes for diagnostic angiogram patients and 5 minutes for anti-coagulated patients undergoing a percutaneous coronary intervention (PCI). Most of the patients were able to sit up within 15-30 minutes and were able to walk in about one hour after an angiogram and two hours after a PCI. Investigators found no major complications related to use of the Arstasis device.
A small histological study by Patel et al involved examining bovine arteries following AXERA® and control (Seldinger access) arteriotomy and indicated that healing after AXERA® access was similar to Seldinger access technique with no evidence of dissection or pseudoaneurysm [6969. Patel R, Virmani R, Kolodgie FD, Cogan D. TCT-797 Vascular healing after AXERA(tm) Access and Closure: a 30 day histopathological assessment in the ovine model. Journal of the American College of Cardiology. 2013;62(18 Supplement 1):B242. ].
EXTRAVASCULAR MANUAL COMPRESSION
Extravascular manual compression, with or without the potentially augmenting effect of haemostasis patches, remains the gold standard of vascular sheath removal. The technique involves applying digital pressure proximal to the skin puncture point in order to compress the femoral artery and surrounding soft tissues against the femoral head. Vertical pressure through a straight upper limb reduces the operator fatigue that can be encountered. Compression should be applied such that haemostasis is achieved with a palpable pulse. If this is difficult to achieve (e.g., because of large body habitus), then sufficient compression can be applied to obliterate temporarily the femoral pulse (10 minutes on/off). Some operators reduce the blood pressure with nitrate medication in significantly hypertensive individuals in order to facilitate haemostasis.
An arterial puncture, which is either proximal or distal to the femoral head, means manual compression will generate less pressure over the puncture point and hence be less successful in achieving optimal haemostasis.
Although manual pressure is widely used, especially for smaller diagnostic sheaths, the technique is inexact and often stressful to the patient and operator, should haemostasis be difficult to achieve. Interventional procedures usually require the use of larger sheath sizes and are performed in combination with an anticoagulation regime. For these patients, once the ACT has dropped to a satisfactory level, manual compression can be used to control bleeding. Therefore, early sheath removal is precluded and bed rest of a variable duration is necessary, dependent on the level of anticoagulation. These circumstances may exist with excessive bleeding, intra-aortic balloon pump (IABP) removal, or arterial insufficiency.
The use of a mechanical compression device in these and many other scenarios can be beneficial in achieving primary haemostasis with minimum discomfort to the patient and at a lower cost than a closure device.
In addition, for certain high risk patients who have received aggressive anticoagulation therapy, indwelling sheaths have to be left in situ for several hours or overnight and compression must be maintained for an extended period of time after sheath removal. Prolonged pressure bandage is also a source of discomfort and hazard since the development of complications such as haematoma, oozing and arterial bleeding are concealed and noticed much too late.
It should be noted that there are several situations where femoral anatomy is non-ideal (a small calibre vessel < 5 mm, a calcified artery by fluoroscopy, PVD up to 70% stenosis, and arteriotomy site at or below the bifurcation in diagnostic catheterisation procedures) and which preclude use of a vascular closure device. In these circumstances, manual compression should remain the closure method of choice.
FEMOSTOP™ COMPRESSION DEVICE
The FemoStop™ Gold Femoral Compression System (Abbott Vascular, Santa Clara, CA, USA) is indicated for use in the compression of the femoral artery or vein after vessel cannulation, and in ultrasound guided compression repair after femoral artery pseudoaneurysm. This device is, however, contraindicated in patients with severe peripheral vascular disease.
The system consists of an arch with a sterile pneumatic pressure dome, a belt and reusable pump with manometer ( Figure 24 ). The pressure dome is placed over the vessel puncture site in the groin. The belt is placed around the patient. The dome, when inflated, applies a mechanical pressure over the vessel puncture site to induce haemostasis. The reusable pump and the manometer control the pressure of the dome and allow the application of the desired pressure to maximise haemostasis. The arch and the belt provide counter-pressure for the dome. The transparency of the dome allows for total puncture visibility. Importantly, the point of maximal pressure identified at the apex of the dome must be applied proximal to the femoral artery puncture. A potential error is to assume that the skin incision/puncture point represents the femoral arterial puncture point.
Trial data for FemoStopTM
Clinical studies of the use of passive devices are limited especially as the FemoStop™ has been used primarily as the control arm in studies after endovascular procedures [2828. Amin FR, Yousufuddin M, Stables R, Shamim W, Al-Nasser F, Coats AJS, et al. Femoral haemostasis after transcatheter therapeutic intervention: a prospective randomised study of the angio-seal device vs the femostop device. International Journal of Cardiology. 2000;76(2):235-40. ]. A small randomised study found no differences in groin complications with FemoStop when compared with manual compression [7070. Rudisill PT, Williams LB, Craig S, Schopp P. Study of mechanical versus manual/mechanical compression following various interventional cardiology procedures. J Cardiovasc Nurs. 1997;11(2):15-21. ]. By contrast, a randomised study which enrolled 212 patients showed FemoStop use to be associated with an increased rate of vascular complications when compared with manual compression [7171. Walker SB, Cleary S, Higgins M. Comparison of the FemoStop device and manual pressure in reducing groin puncture site complications following coronary angioplasty and coronary stent placement. International Journal of Nursing Practice. 2001;7(6):366-75. ].
SAFEGUARD®
The Safeguard® (Maquet Cardiovascular [Datascope Corp.], Fairfield, NJ, USA) device is a latex-free sterile pressure-assisted dressing which is designed to maintain and protect haemostasis while providing the ability to monitor clearly the access site. Safeguard® is a single-use adhesive dressing with a transparent inflatable plastic bulb ( Figure 25 ). When inflated, the bulb is inelastic and provides consistent pressure over the wound site to maintain haemostasis. The see-through bulb allows easy monitoring of the wound site. The Safeguard® is available in 12cm and 24cm sizes.
HAEMOSTASIS PADS
Haemostasis pads contain pro-coagulant coatings that serve to enhance the coagulation pathway with the aim of achieving quicker vascular repair. Several devices are available on the market including the Clo-Sur P.A.D® (Merit Medical Systems, Inc., South Jordan, UT, US), D-stat® Dry (Vascular solutions, MN, US), Neptune Pad (TZ Medical Inc., Portland, OR, US) and SyvekExcel® Patches (Marine polymer technologies Inc, Dankers, MS, US). Their mode of action is illustrated by the Clo-Sur P.A.D® below.
CLO-SUR P.A.D®
The Clo-Sur P.A.D® (pressure applied dressing) (Merit Medical Systems, Inc., South Jordan, UT, US) device is a soft, non-woven hydrophilic wound dressing. This device consists of the hydrophilic naturally occurring biopolymer Polyprolate™ acetate. This linear biopolymer is cationically charged in its dry state. It is the chain of positive charges which endows Polyprolate™ with its blood-coagulant properties. Cationic Polyprolate™ forms a coagulum with heparinised blood, defibrinated blood and washed red blood cells.
- The Clo-Sur P.A.D® procedure
- Proximal pressure is held above the puncture site and the sheath is removed.
- The Clo-Sur P.A.D® is placed over the puncture site and proximal pressure is released.
- A small amount of blood is allowed to contact the Clo-Sur P.A.D®.
- Constant pressure is then held for a minimum of 10 minutes or until haemostasis is achieved.
- A dressing is applied over the Clo-Sur P.A.D®.
- The dressing is removed after 24 hours. The Clo-Sur P.A.D® dissolves when dampened with water
Trial data for Haemostasis pads
Studies of haemostasis pads as an adjunctive technique to manual compression compared to compression alone have generally demonstrated limited efficacy with similar time to haemostasis, vascular complications rate and bed-rest time [7272. Nguyen N, Hasan S, Caufield L, Ling FS, Narins CR. Randomized controlled trial of topical hemostasis pad use for achieving vascular hemostasis following percutaneous coronary intervention. Catheterization and Cardiovascular Interventions. 2007;69(6):801-7. , 7373. Nader RG, Garcia JC, Drushal K, Pesek T. Clinical evaluation of SyvekPatch in patients undergoing interventional, EPS and diagnostic cardiac catheterization procedures. J Invasive Cardiol. 2002;14(6):305-7. ]. There is also a high technical failure rate associated with these devices [7474. Mlekusch W, Dick P, Haumer M, Sabeti S, Minar E, Schillinger M. Arterial Puncture Site Management after Percutaneous Transluminal Procedures Using a Hemostatic Wound Dressing (Clo-Sur PAD) versus Conventional Manual Compression: A Randomized Controlled Trial. Journal of Endovascular Therapy. 2006;13(1):23-31. ].
The largest analysis of these devices comes from the National Cardiovascular Data Registry, which showed that their use is associated with a slightly smaller risk of bleeding, or vascular complications compared to conventional manual compression [5757. Tavris DR, Wang Y, Jacobs S, Gallauresi B, Curtis J, Messenger J, et al. Bleeding and vascular complications at the femoral access site following percutaneous coronary intervention (PCI): an evaluation of hemostasis strategies. J Invasive Cardiol. 2012;24(7):328-34. ].
LARGE-BORE ACCESS and VCDs
The topic of vascular closure following TAVIs is covered in chapter 3.32 but it is worth briefly mentioning some of the devices available for the closure of large arteriotomies. Structural heart procedures such as TAVI and the implantation of mechanical circulatory support devices (e.g Impella) require large bore sheaths (between 14-20Fr). Larger arteriotomies are associated with greater morbidity including higher bleeding risk, delayed ambulation as well as higher cost of care. Surgical closure offers the best control of bleeding but is associated with a higher incidence of infection and need for debridement [4141. Torsello GB, Kasprzak B, Klenk E, Tessarek J, Osada N, Torsello GF. Endovascular suture versus cutdown for endovascular aneurysm repair: a prospective randomized pilot study1. Journal of Vascular Surgery. 2003;38(1):78-82. ].
Closure of large bore access sites can be challenging using conventional VCDs. The currently widely utilised technique is that of the preclosure with either the Prostar XL and the Proglide VCD. This is done by partially deploying the sutures after the artery is accessed by a small calibre sheath and then replacing the smaller sheath with the large-bore arterial sheath ( Video 1 ). After completion of the procedure, the sheath is removed and the sutures tied in the standard manner. However this technique is associated with a not inconsequential bleeding rate [7575. Van Mieghem NM, Latib A, van der Heyden J, van Gils L, Daemen J, Sorzano T, et al. Percutaneous Plug-Based Arteriotomy Closure Device for Large-Bore Access: A Multicenter Prospective Study. JACC: Cardiovascular Interventions. 2017;10(6):613-9. ]. A retrospective analysis of 472 propensity matched patients undergoing transfemoral TAVI showed that the Prostar VCD was associated with a higher major vascular complication rate [4545. Barbash IM, Barbanti M, Webb J, Molina-Martin De Nicolas J, Abramowitz Y, Latib A, et al. Comparison of vascular closure devices for access site closure after transfemoral aortic valve implantation. European Heart Journal. 2015;36(47):3370-9. ].
The exponential growth of TAVI over the past decade has spurred interest in the development of dedicated VCDs for large bore arteriotomy closure. Three of the devices available on the market are briefly outlined below.
MANTA™ device
The MANTA™ device (Essential Medical Inc, Malvern, Pennsylvania) has CE mark to close puncture sizes ranging from 10 to 25 French sheaths. The concept is similar to that of the Angio-seal. The device uses a collagen implant which is secured by an intra-luminal anchor that allows closure of large-bore arteriotomies ( Figure 26 ). The device is available in two sizes – 14 and 18Fr which close 10-14 Fr and 15-22 sites respectively.
PerQseal® device
The PerQseal® device (Vivasure Medical Ltd, Galway, IR) is a fully bioresorbable device indicated for closure of TAVI access sites. The device consists of a polymer implant which is deployed inside the vessel wall ( Figure 27 ). The implant has a flexible intra-vascular patch and a supporting scaffold. Its abluminal surface is textured to promote adherence of the patch to the vessel wall. Part of the scaffold extends through the arteriotomy and includes a locator which helps maintain the implant in position. After deployment, the implant is endothelialised and fully absorbed.
Inseal VCD
The Inseal VCD (Inseal Medical Ltd, Caesarea, Israel) is indicated for closure of punctures ranging from 14-21Fr sizes. The device consists of a self-expandable Nitinol frame covered by a biodegradable membrane. This membrane covers the puncture site and is maintained in place by the blood pressure ( Figure 28 ). This system is delivered through the same sheath used for the large bore procedure.
RADIAL ACCESS
Diagnostic and interventional cardiovascular procedures are routinely performed via the common femoral or the radial artery. The transfemoral route initially dominated the interventional landscape but has largely been superseded by the radial approach in many countries in Europe and Asia. While the transfemoral route is still widely used in United States, data from the National Cardiovascular Data Registry shows a growing adoption of the radial approach [66. Masoudi FA, Ponirakis A, de Lemos JA, Jollis JG, Kremers M, Messenger JC, et al. Trends in US Cardiovascular Care: 2016 Report From 4 ACC National Cardiovascular Data Registries. Journal of the American College of Cardiology. 2017;69(11):1427-50. ].
Transradial access is associated with fewer bleeding and vascular complications [7676. Valgimigli M, Frigoli E, Leonardi S, Vranckx P, Rothenbühler M, Tebaldi M, et al. Radial versus femoral access and bivalirudin versus unfractionated heparin in invasively managed patients with acute coronary syndrome (MATRIX): final 1-year results of a multicentre, randomised controlled trial. The Lancet. 2018;392(10150):835-48. , 7777. Jolly SS, Yusuf S, Cairns J, Niemelä K, Xavier D, Widimsky P, et al. Radial versus femoral access for coronary angiography and intervention in patients with acute coronary syndromes (RIVAL): a randomised, parallel group, multicentre trial. The Lancet. 2011;377(9775):1409-20. ] as well as allowing earlier patient ambulation. Most of the disadvantages of the femoral technique are non-existent in the transradial approach. Even in obese patients, the radial artery is close to the skin surface, making the initial needle puncture simple and straightforward. For the same reason, when the procedure has been completed, a short compression of the radial artery can stop the bleeding, even when the patient has been aggressively anticoagulated. Should any bleeding occur, it can be seen immediately. Finally, unlike the proximity of the femoral artery to the femoral nerve, the radial artery is not close to a major nerve, so the likelihood of nerve injury during the procedure is very low. A meta-analysis of four randomized and seven non-randomized studies, which compared the radial approach with the femoral approach plus achievement of haemostasis with VCD, showed a reduction in both access site complications and major bleeding when the radial approach was used [7878. Rigattieri S, Sciahbasi A, Alonzo A, Nolan J, Cox N, Chodór P, et al. TCT-421 Radial Approach versus Femoral Approach with Vascular Closure Devices: Systematic Review and Meta-analysis. Journal of the American College of Cardiology. 2015;66(15 Supplement):B171. ]. Recently, a new technique of accessing the distal radial artery via the anatomical snuffbox has been demonstrated [7979. Kiemeneij F. Left distal transradial access in the anatomical snuffbox for coronary angiography (ldTRA) and interventions (ldTRI). EuroIntervention. 2017;13(7):851-7. ]. This should help mitigate the risk of radial artery occlusion proximal to the wrist.
In addition to lower complication rates, another advantage of the radial technique is that patients do not need to lie flat and still for 4-6 hours, or experience what is sometimes a painful manual compression of the artery. Patients leave the catheterisation lab and are able to sit up and walk almost immediately. Because of the simpler healing process for the arterial puncture in the wrist, patients may also be discharged home without having to spend the night in hospital.
The 2018 ESC/EACTS guidelines on myocardial revascularisation recommends the radial approach as the standard access of choice for PCI unless not technically feasible [8080. Neumann F-J, Sousa-Uva M, Ahlsson A, Alfonso F, Banning AP, Benedetto U, et al. 2018 ESC/EACTS Guidelines on myocardial revascularization. European Heart Journal. 2018:ehy394-ehy. ]. Similarly, a recent position statement from the AHA recommends the transradial approach as the default strategy in patients presenting with ACS [8181. Mason Peter J, Shah B, Tamis-Holland Jacqueline E, Bittl John A, Cohen Mauricio G, Safirstein J, et al. An Update on Radial Artery Access and Best Practices for Transradial Coronary Angiography and Intervention in Acute Coronary Syndrome: A Scientific Statement From the American Heart Association. Circulation: Cardiovascular Interventions. 2018;11(9):e000035. ].
TRANSRADIAL COMPRESSION DEVICES
As the use of the transradial access has increased, so has the demand for compression devices to address this growing need. Manual pressure with gauze were initially replaced by rigid wrist bands such as Hemoband. Nowadays, these have largely been superseded by air filled bracelets.
A number of devices have been designed to provide haemostasis, from a simple, plastic haemodialysis band compressing a gauze bullet over the radial arterial puncture to specifically designed compression bands ( Table 4 ). Topical haemostatic agents are also available for use in conjunction with radial bands in order to shorten the time to achieve haemostasis. A wide variety of compounds are available for this purpose.
Regardless of the chosen device, there are a few critical steps in this process. After removing the drapes from the hand and arm, begin sheath removal as noted below.
- Position the device loosely around the wrist prior to sheath removal with the compression portion of the device covering the arteriotomy site.
- A vasodilator (e.g., verapamil 2.5 mg) can be given through the sheath to minimise spasm during sheath removal.
- Remove the sheath slowly and smoothly while slowly tightening the selected compression method as per the device IFU. One common mistake is to tighten the device too aggressively before the sheath is completely removed, leading to patient discomfort. For most pressure bands, removal of the sheath and tightening of the band should be simultaneous, both for patient comfort and for adequate haemostasis.
- Check oximetric finger (preferably the thumb) arterial flow with the closure device in place. Flow through the radial artery is thought to increase the likelihood of post procedure radial artery patency.
- Check the patient in 1 hour. Loosen the device slightly as per the particular IFUs. The patient can be discharged after 2 hours (if conscious sedation discharge parameters are met). If there is re-bleeding, reapply pressure and return to Step 5.
The patient is given instructions about puncture site compression with the fingers if (rare) late bleeding occurs.
Two of the commonly used compression devices are outlined below.
TR BAND®
This is a radial artery compression device designed to stop bleeding safely and comfortably after transradial procedures ( Figure 29A and Figure 29B ) The TR Band® (Terumo Medical Corp., Somerset, NJ, USA) is an easy-to-apply transparent band with precision pressure balloons which enable unobstructed visualisation and monitoring of radial artery compression.
The primary benefit that the TR Band® has over similar devices is its directed radial compression, which enables haemostasis achievement without compromising local nerve structure. The TR Band® does so with its two separate inflatable balloons. A large balloon compresses the entire puncture site, and a small balloon gives it an angled direction for point compression. This dual system enables the TR Band® to avoid compression of the nerves or the ulnar artery, which can cause discomfort and/or complications. The transparency of the band, which allows full visibility to the clinician during the critical post-procedure stage, is another significant clinical benefit of this product.
HELIX™
The Helix™ device (Vascular perspectives) allows a controlled compression of the radial artery ( Figure 30 ). The aim is to improve radial patency rate post procedure. The design features a screw-bolt on top of the transparent silicone pressure pad. The steps are outlined below:
- Withdraw the sheath by approximately 2-3cm to allow placement of the device
- Orientate the device so that the M logo faces the little finger. Position the device so that the centre of the cushion is placed 5mm behind the puncture site. The puncture site should be located slightly distal to the screw-bolt section and as close to the middle of the silicone pad as possible.
- Strap the device firmly with the band below the wrist pad to ensure that it does not move during this process.
- Rotate the turn cap clockwise until the green mark is visible. In case of hypertension, continue rotation until the yellow top appears.
- Remove the sheath. If bleeding occurs, tighten the device by rotating the turn cap until bleeding stops.
- Apply a pulse oximeter over the index finger. A normal waveform should be visible.
- Occlude the ulnar artery and inspect the waveform of the pulse oximeter. If there is no reading, then gradually reduce loosen the device until a normal waveform re-appears while maintaining ulnar occlusion. Haemostasis is achieved.
Complications with VCD use
VCD pivotal studies have generally included highly selected, low-risk patients in studies with small sample sizes, with 250 to 600 patients. Complication rates of 3-5% have been reported with use of VCDs [1717. Baim DS, Knopf WD, Hinohara T, Schwarten DE, Schatz RA, Pinkerton CA, et al. Suture-mediated closure of the femoral access site after cardiac catheterization: results of the suture to ambulate aNd discharge (STAND I and STAND ii) trials. American Journal of Cardiology. 2000;85(7):864-9. , 1919. Sanborn TA, Gibbs HH, Brinker JA, Knopf WD, Kosinski EJ, Roubin GS. A multicenter randomized trial comparing a percutaneous collagen hemotasis device with conventional manual compression after diagnostic angiography and angioplasty. Journal of the American College of Cardiology. 1993;22(5):1273-9. , 2020. Hermiller JB, Simonton C, Hinohara T, Lee D, Cannon L, Mooney M, et al. The StarClose® vascular closure system: Interventional results from the CLIP study. Catheterization and Cardiovascular Interventions. 2006;68(5):677-83. , 8282. Dangas G, Mehran R, Kokolis S, Feldman D, Satler LF, Pichard AD, et al. Vascular complications after percutaneous coronary interventions following hemostasis with manual compression versus arteriotomy closure devices. Journal of the American College of Cardiology. 2001;38(3):638-41. ]. Without the inclusion of higher risk patients, there is a multitude of precautions and contraindications for each device ( Table 5 )
There have been many case reports of rare complications occurring with VCDs such as infections, arterial laceration, uncontrolled bleeding, pseudoaneurysm, arteriovenous fistula, device embolism and limb ischaemia [1111. Piper WD, Malenka DJ, Ryan TJ, Shubrooks SJ, O’Connor GT, Robb JF, et al. Predicting vascular complications in percutaneous coronary interventions. American Heart Journal. 2003;145(6):1022-9. , 1212. Tavris DR, Gallauresi BA, Lin B, Rich SE, Shaw RE, Weintraub WS, et al. Risk of local adverse events following cardiac catheterization by hemostasis device use and gender. J Invasive Cardiol. 2004;16(9):459-64. ]. Whilst complications are rare, they can be devastating, resulting in either surgical repair, often under urgent circumstances, or even limb loss. These data underline the importance of meticulous attention both to the suitability of a femoral puncture for device closure and to the technique itself, which requires formal supervised training. Any persisting discomfort following device deployment should prompt a thorough examination and imaging to exclude complications.
As a result of many adverse events post-VCD deployment, the FDA initiated a study of 166,680 patients via the ACC-NCDR database “to assess the relative risk of complications after the use of the 2 main types of haemostasis device” [1313. Tavris D, Gross T, Gallauresi B, Kessler L. Arteriotomy closure devices—the FDA perspective - Editorials published in the Journal of the American College of Cardiologyreflect the views of the authors and do not necessarily represent the views of JACCor the American College of Cardiology. Journal of the American College of Cardiology. 2001;38(3):642-4. ]. However, concerns that use of VCDs led to worse outcomes than manual compression have not been supported by multicentre registries and meta-analyses.
DATA
Trial and registries
Given the availability of many randomised studies with conflicting safety data and multiple limitations, it has become important to consider the evidence from larger registries and databases regarding the efficacy and safety of the devices as well as their comparisons against manual compression and against each other. Some of the larger trials and registry data are outlined below.
A large single-centred retrospective analysis [8282. Dangas G, Mehran R, Kokolis S, Feldman D, Satler LF, Pichard AD, et al. Vascular complications after percutaneous coronary interventions following hemostasis with manual compression versus arteriotomy closure devices. Journal of the American College of Cardiology. 2001;38(3):638-41. ] compared vascular outcomes in almost 6,000 patients treated with manual compression with 516 VCD uses (371 Angio-Seal, 101 Techstar [suture-based]) after diagnostic and interventional procedures. This demonstrated that the use of VCDs was associated with significantly more haematomas, a larger drop in haematocrit and a greater need for vascular surgical repair of the access site. Multivariate analysis was performed and clarified that age, body surface area and sex, not the use of VCDs, were independent predictors of vascular complications.
The Acuity trial compared access site bleeding in 11,621 patients undergoing diagnostic and interventional procedures. 4,307 of these patients (37.1%) received a VCD [2121. Sanborn Timothy A, Ebrahimi R, Manoukian Steven V, McLaurin Brent T, Cox David A, Feit F, et al. Impact of Femoral Vascular Closure Devices and Antithrombotic Therapy on Access Site Bleeding in Acute Coronary Syndromes. Circulation: Cardiovascular Interventions. 2010;3(1):57-62. ]. Rates of access site bleeding were lower with VCD compared with no VCD (2.5% vs. 3.3%, p=0.01). Stepwise logistic regression revealed that a VCD was an independent determinant of freedom from major access site bleeding. The ISAR-CLOSURE trial compared a VCD-based strategy (with Femoseal or Exoseal) against manual compression to close arteriotomies after diagnositic coronary angiography in 3015 patients [8383. Schulz-Schüpke S, Helde S, Gewalt S, et al. Comparison of vascular closure devices vs manual compression after femoral artery puncture: The isar-closure randomized clinical trial. JAMA. 2014;312(19):1981-7. ]. The primary endpoint of access site-related vascular complications at 30 days was similar in both groups (6.9% with VCD and 7.9% with compression).
Angio-Seal and Perclose were compared with manual compression in 1,500 patients with a finding that Angio-Seal was associated with a shorter time to haemostasis and ambulation than Perclose [2222. Duffin DC, Muhlestein JB, Allisson SB, Horne BD, Fowles RE, Sorensen SG, et al. Femoral arterial puncture management after percutaneous coronary procedures: a comparison of clinical outcomes and patient satisfaction between manual compression and two different vascular closure devices. J Invasive Cardiol. 2001;13(5):354-62. Epub 2001/06/01. ]. Perclose use was associated with more access-site complications than Angio-Seal and manual compression. The Angio-Seal and Perclose devices were again compared with manual compression in a registry (84) of patients undergoing PCI with abciximab in which 524 patients used the Angio-Seal, 2177 the Perclose, and 1824 underwent manual compression. Haemostasis was significantly improved in the Angio-Seal group versus the Perclose group (p<0.05) and overall complications were reduced among patients with successful VCD use compared with manual compression (p<0.05). A single centre retrospective study compared five different closure devices (Angioseal, Perclose, Starclose, Exoseal and Mynx) used in an outpatient endovascular surgery centre [8585. Jones LE, Yang KH, Feldtman RW, Uceda PV, Ferrara CA, Caruso JM, et al. Safety and Efficacy of Arterial Closure Devices in an Office-Based Angiosuite. Annals of Vascular Surgery. 2018;51:10-7. ]. Femoral arterial closure with closure devices was performed in 1810 patients. The Angioseal device had the smallest rate of failure (3.5%) and minor complications (1.3%).
The British Cardiovascular Interventional Society (BCIS) examined the 30 day mortality between manual pressure and VCD in the UK registry of 271845 patients who had undergone PCI between 2006 and 2011 [8686. Farooq V, Goedhart D, Ludman P, de Belder Mark A, Harcombe A, El-Omar M. Relationship Between Femoral Vascular Closure Devices and Short-Term Mortality From 271 845 Percutaneous Coronary Intervention Procedures Performed in the United Kingdom Between 2006 and 2011. Circulation: Cardiovascular Interventions. 2016;9(6):e003560. ]. After propensity matching, the use of VCDs was found to be associated with a small but statistically significant benefit in 30 day mortality (1.8% v 2.0%, p<0.001). This effect was more pronounced in females and acute presentations (ACS).
Data from the large ACC-NCDR registry has been used to compare VCDs versus manual compression. In an analysis of 166,680 patients across 214 sites, the risk of vascular complications was lower in men than in women, and after diagnostic rather than interventional procedures when a VCD was used(12). Multivariate analysis demonstrated VCD use to be associated with a lower risk of any vascular complication, driven predominantly by lower rates of bleeding and pseudoaneurysm formation in diagnostic procedures only. In a more recent analysis of the same registry, a total of 2056585 cases were examined with just over half of the cases (51.2%) having received a VCD [8787. Wimmer Neil J, Secemsky Eric A, Mauri L, Roe Matthew T, Saha-Chaudhuri P, Dai D, et al. Effectiveness of Arterial Closure Devices for Preventing Complications With Percutaneous Coronary Intervention. Circulation: Cardiovascular Interventions. 2016;9(4):e003464. ]. Global vascular access site complication rate was 1.5%. The use of VCD was associated with a 0.36% absolute risk reduction in vascular access complications compared with manual compression.
The overall non-randomised observational data appears to conclude that VCDs are efficacious but does not demonstrate definitively their safety versus manual compression or versus each other.
META-ANALYSES
( Table 6 )
Several meta-analysis have been conducted comparing VCDs with manual compression and are briefly discussed below.
Koreny et al looked at data assessing the efficacy and safety of VCDs compared with manual compression in 30 randomised trials enrolling 4,000 patients. Overall time to haemostasis was reduced by a mean of 17 minutes with VCDs. There were non-significant trends towards increased haematomas, bleeding, arteriovenous fistulas and pseudoaneurysms with VCDs [1010. Koreny M, Riedmüller E, Nikfardjam M, Siostrzonek P, Müllner M. Arterial puncture closing devices compared with standard manual compression after cardiac catheterization: Systematic review and meta-analysis. JAMA. 2004;291(3):350-7. ].
Nikolsky et al analysed the safety of VCDs versus manual compression in a meta-analysis of 37,066 patients in 30 centres [1515. Nikolsky E, Mehran R, Halkin A, Aymong ED, Mintz GS, Lasic Z, et al. Vascular complications associated with arteriotomy closure devices in patients undergoing percutaneous coronary procedures: A meta-analysis. Journal of the American College of Cardiology. 2004;44(6):1200-9. ]. In patients undergoing PCI, there was an increased rate of vascular complications (OR 1.34, 95% CI 1.01 to 1.79) when compared with manual compression. However, in patients having only a diagnostic procedure, there was a non-significant trend which favoured VCDs over manual compression. When the devices were compared, the Angio-Seal and Perclose devices had comparable results to manual compression; however, the VasoSeal was associated with increased vascular complications compared with manual compression (OR 2.27, 95% CI 1.35 to 3.80).
Vaitkus et al pooled data from 16 randomised controlled trials with 5,084 patients and showed an overall reduction in vascular complications with VCD use (OR 0.89, 95% CI 0.86 to 0.91) [1616. Vaitkus PT. A meta-analysis of percutaneous vascular closure devices after diagnostic catheterization and percutaneous coronary intervention. J Invasive Cardiol. 2004;16(5):243-6. ], in particular with the Angio-Seal and Perclose devices. The VasoSeal device was associated with a higher risk of vascular complications. When they looked at PCI procedures only, there was a significant reduction in vascular complications with the Angio-Seal (OR 0.51, 95% CI 0.45 to 0.58), no difference with Perclose (OR 1.0, 95% CI 0.13 to 7.48) and an increased risk with VasoSeal (OR 1.18, 95% CI 1.16 to 1.20).
Another meta-analysis evaluated the safety and efficacy of VCDs in 7,528 patients in 31 prospective randomised studies [8888. Biancari F, D’Andrea V, Marco CD, Savino G, Tiozzo V, Catania A. Meta-analysis of randomized trials on the efficacy of vascular closure devices after diagnostic angiography and angioplasty. American Heart Journal. 2010;159(4):518-31. ]. It is worth noting that most of these studies excluded patients at high risk of puncture site complications. There was no overall difference in terms of groin haematoma, bleeding, pseudoaneurysm and blood transfusion. The use of VCDs was associated with an increased risk of infection (p=0.02), lower limb ischaemia/arterial stenosis/device entrapment in the artery (p=0.07) and need of vascular surgery for arterial complications (p=0.10). However, the use of VCDs was associated with a significantly shorter time to haemostasis and maybe a shorter recovery time.
Cox et al. conducted a systematic review of 34 randomised controlled trials (RCT) comparing vascular closure devices to manual compression [8989. Cox T, Blair L, Huntington C, Lincourt A, Sing R, Heniford BT. Systematic Review of Randomized Controlled Trials Comparing Manual Compression to Vascular Closure Devices for Diagnostic and Therapeutic Arterial Procedures. Surg Technol Int. 2015;27:32-44. ]. The use of VCDs shortened time to haemostasis, allowed more rapid ambulation and earlier discharge compared to manual compression.
A recently published meta-analysis by Iannaccone et al showed that VCDs did not result in fewer vascular complications compared to manual compression (90). However they did have a shorter time to haemostasis. In terms of specific devices, the StarClose had the lowest vascular complication rate while the Angioseal device had the shortest time to haemostasis.
A Cochrane review of 52 randomised controlled trials conducted by Roberson et al. showed that both metal clip-based and suture-based VCDs achieved haemostasis more quickly compared to extrinsic compression [9191. Robertson L, Andras A, Colgan F, Jackson R. Vascular closure devices for femoral arterial puncture site haemostasis. Cochrane Database of Systematic Reviews. 2016(3). ]. Mortality and vascular injury rates between VCDs and extrinsic compression were comparable. The safety and efficacy profiles of the different devices were comparable.
It is difficult to make accurate evaluations as to the efficacy and safety of VCDs from the observational registry data and meta-analyses. The randomised trials are small and appear to have been conducted in patients with lower risk than the current clinical population. These small trials may not accurately identify the different clinical effects of the devices. In addition, the study endpoints are variable with some studies having a composite endpoint [1212. Tavris DR, Gallauresi BA, Lin B, Rich SE, Shaw RE, Weintraub WS, et al. Risk of local adverse events following cardiac catheterization by hemostasis device use and gender. J Invasive Cardiol. 2004;16(9):459-64. , 8282. Dangas G, Mehran R, Kokolis S, Feldman D, Satler LF, Pichard AD, et al. Vascular complications after percutaneous coronary interventions following hemostasis with manual compression versus arteriotomy closure devices. Journal of the American College of Cardiology. 2001;38(3):638-41. ], some using a single endpoint [1010. Koreny M, Riedmüller E, Nikfardjam M, Siostrzonek P, Müllner M. Arterial puncture closing devices compared with standard manual compression after cardiac catheterization: Systematic review and meta-analysis. JAMA. 2004;291(3):350-7. , 2323. Chevalier B, Lancelin B, Koning R, Henry M, Gommeaux A, Pilliere R, et al. Effect of a closure device on complication rates in high-local-risk patients: Results of a randomized multicenter trial. Catheterization and Cardiovascular Interventions. 2003;58(3):285-91. ], and some using a different definition of “vascular complications”.
Taking all this into account, there are several conclusions which may be drawn:
- In patients undergoing diagnostic cardiac catheterisation there is a 0.5% to 1.7% rate of vascular complications [1010. Koreny M, Riedmüller E, Nikfardjam M, Siostrzonek P, Müllner M. Arterial puncture closing devices compared with standard manual compression after cardiac catheterization: Systematic review and meta-analysis. JAMA. 2004;291(3):350-7. , 1212. Tavris DR, Gallauresi BA, Lin B, Rich SE, Shaw RE, Weintraub WS, et al. Risk of local adverse events following cardiac catheterization by hemostasis device use and gender. J Invasive Cardiol. 2004;16(9):459-64. ], which is not consistently altered by the use of VCDs across all the studies. The largest study from the ACC-NCDR suggested a significant decrease in complications with VCD usage compared with manual compression [8787. Wimmer Neil J, Secemsky Eric A, Mauri L, Roe Matthew T, Saha-Chaudhuri P, Dai D, et al. Effectiveness of Arterial Closure Devices for Preventing Complications With Percutaneous Coronary Intervention. Circulation: Cardiovascular Interventions. 2016;9(4):e003464. ]. It is possible that this reduction in complications is directly related to the VCD or alternatively that it reflects a reluctance by operators to use VCDs in patients who have already had a complication at the end of the diagnostic procedure or in circumstances where both VCD use and manual compression have been associated with increased complications [1414. Dauerman HL, Applegate RJ, Cohen DJ. Vascular Closure Devices: The Second Decade. Journal of the American College of Cardiology. 2007;50(17):1617-26. ].
- Among patients undergoing PCI there is a 0.8% to 5.5% rate of vascular complications. There is no data to suggest clearly an increased risk of vascular complications with VCD use [1212. Tavris DR, Gallauresi BA, Lin B, Rich SE, Shaw RE, Weintraub WS, et al. Risk of local adverse events following cardiac catheterization by hemostasis device use and gender. J Invasive Cardiol. 2004;16(9):459-64. , 1515. Nikolsky E, Mehran R, Halkin A, Aymong ED, Mintz GS, Lasic Z, et al. Vascular complications associated with arteriotomy closure devices in patients undergoing percutaneous coronary procedures: A meta-analysis. Journal of the American College of Cardiology. 2004;44(6):1200-9. , 1616. Vaitkus PT. A meta-analysis of percutaneous vascular closure devices after diagnostic catheterization and percutaneous coronary intervention. J Invasive Cardiol. 2004;16(5):243-6. ]: some studies suggest that VCDs decrease complications compared with manual compression [1616. Vaitkus PT. A meta-analysis of percutaneous vascular closure devices after diagnostic catheterization and percutaneous coronary intervention. J Invasive Cardiol. 2004;16(5):243-6. , 2323. Chevalier B, Lancelin B, Koning R, Henry M, Gommeaux A, Pilliere R, et al. Effect of a closure device on complication rates in high-local-risk patients: Results of a randomized multicenter trial. Catheterization and Cardiovascular Interventions. 2003;58(3):285-91. ], some studies suggest a potentially increased risk with VCDs, and some suggest complication rates are similar [1515. Nikolsky E, Mehran R, Halkin A, Aymong ED, Mintz GS, Lasic Z, et al. Vascular complications associated with arteriotomy closure devices in patients undergoing percutaneous coronary procedures: A meta-analysis. Journal of the American College of Cardiology. 2004;44(6):1200-9. , 9292. Applegate RJ, Sacrinty MT, Kutcher MA, Baki TT, Gandhi SK, Santos RM, et al. Propensity score analysis of vascular complications after diagnostic cardiac catheterization and percutaneous coronary intervention using thrombin hemostatic patch-facilitated manual compression. J Invasive Cardiol. 2007;19(4):164-70. ].






















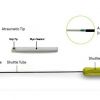





























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}